Dear MNE users,
I am new to MNE software. I wish to establish source space based on the
brain surface. I first run recon-all in freesurfer (latest version) to
generate potentially necessary files for MNE. Then I run
Dear MNE users,
I am new to MNE software. I wish to establish source space based on the
brain surface. I first run recon-all in freesurfer (latest version) to
generate potentially necessary files for MNE. Then I run
hi Peng,
I am new to MNE software. I wish to establish source space based on the
brain surface. I first run recon-all in freesurfer (latest version) to
generate potentially necessary files for MNE. Then I runmne_setup_source_space --subject <my subject> --ico 4 --overwrite
to generate the source locations for a particular subject (<my subject>).
Then I had many files in the /bem folder, including an xxx-rh.pnt file,
which I assume is the coordinates of these locations in the right
hemisphere.
extension of source spaces is "-src.fif"
a source space is defined relatively to a surface so it's not common
to try to visualize it with freeview. I am not surprised you have coordinate
alignment problems. But there is nothing to fix.
basically you're good to go with the next step.
For full script for the anatomical pipeline see for example:
HTH
Alex
Thank you Alex for the reply.
1. I suppose you meant freeview is not the optimal tool for visualizing
source space of mne. However, I wish to check the source space by
visualisation before I move on to source reconstructions. I tried
mne_analyze instead of freeview:
1.1 It seemed to detect surfaces generated by freesurfer (such as inflated
etc.) automatically, which I can load by <load surfaces...> in the menu;
but not those surfaces generated by watershed, even after I copy them to
the <surf> subfolder under the subject folder.
1.2 I cannot load -src.fif either in mne_analyze. It complains 'Come on, It
is not an MEG/EEG measurement file'...
2. I tried the example in the script you provided (watershed), error
happened while Decimating the dense tessellation...
"/Applications/MNE-2.7.4-3378-MacOSX-x86_64/bin/mne_make_scalp_surfaces:
line 125: -nojvm: command not found". I do have <mne_make_scalp_surfaces>
file in the path. (Platform: MacOS10.9.2, macbook pro).
3. If my data are MEG instead of EEG, do I still need to use this skin,
skull, brain segmentations from watershed or mne_setup_source_space on
brain surfaces is already sufficient?
Thank you very much and sorry about so many questions...
Hi Peng,
one thing,
Thank you Alex for the reply.
1. I suppose you meant freeview is not the optimal tool for visualizing
source space of mne. However, I wish to check the source space by
visualisation before I move on to source reconstructions. I tried
mne_analyze instead of freeview:
1.1 It seemed to detect surfaces generated by freesurfer (such as inflated
etc.) automatically, which I can load by <load surfaces...> in the menu;
but not those surfaces generated by watershed, even after I copy them to
the <surf> subfolder under the subject folder.
1.2 I cannot load -src.fif either in mne_analyze. It complains 'Come on,
It is not an MEG/EEG measurement file'...2. I tried the example in the script you provided (watershed), error
happened while Decimating the dense tessellation..."/Applications/MNE-2.7.4-3378-MacOSX-x86_64/bin/mne_make_scalp_surfaces:
line 125: -nojvm: command not found". I do have <mne_make_scalp_surfaces>
file in the path. (Platform: MacOS10.9.2, macbook pro).
that's part of the matlab command invoked. weird. is your Matlab path
specified?
You can also use the python version `mne make_scalp_surfaces` which
produces equivalent decimations.
Denis
3. If my data are MEG instead of EEG, do I still need to use this skin,
skull, brain segmentations from watershed or mne_setup_source_space on
brain surfaces is already sufficient?Thank you very much and sorry about so many questions...
hi Peng,
> I am new to MNE software. I wish to establish source space based on the
> brain surface. I first run recon-all in freesurfer (latest version) to
> generate potentially necessary files for MNE. Then I run
> ===
> mne_setup_source_space --subject <my subject> --ico 4 --overwrite
> ===
> to generate the source locations for a particular subject (<my
>).
>
> Then I had many files in the /bem folder, including an xxx-rh.pnt file,
> which I assume is the coordinates of these locations in the right
> hemisphere.extension of source spaces is "-src.fif"
a source space is defined relatively to a surface so it's not common
to try to visualize it with freeview. I am not surprised you have
coordinate
alignment problems. But there is nothing to fix.basically you're good to go with the next step.
For full script for the anatomical pipeline see for example:
https://github.com/mne-tools/mne-scripts/blob/master/sample-data/run_anatomy_tutorial.sh
HTH
Alex> I then used freeview (part of freesurfer) to load the brain
> image (/mri/brain.mgz) of this subject; and overlap xxx-rh.pnt as dot
sets.
> I surprisingly found these dots are not located one the surface of the
white
> matter as expected, but much lower (several centimeters).
> If I changed <my subject> with fsaverage (template from freesurfer), and
> re-run the above command, the new generated dots seemed to be on the
correct
> place.
> Third, I changed <my subject> with an icbm152 asym template, the result
was
> similar to <my subject> : the dots were lower than the image.
>
> So I am a little confused. Shall I do some coordinates transformation to
> align these dots to the mri image? Or it is just a display problem of
> freeview? I quickly browsed the manual and had not find a direct answer
yet.
> Thanks a lot for any hint!
>
> best
> Peng
>
>
>
> _______________________________________________
> Mne_analysis mailing list
> Mne_analysis at nmr.mgh.harvard.edu
> https://mail.nmr.mgh.harvard.edu/mailman/listinfo/mne_analysis
>
>
> The information in this e-mail is intended only for the person to whom
it is
> addressed. If you believe this e-mail was sent to you in error and the
> contains patient information, please contact the Partners Compliance
> HelpLine at
> http://www.partners.org/complianceline . If the e-mail was sent to you
in
> error
> but does not contain patient information, please contact the sender and
> properly
> dispose of the e-mail.
>
_______________________________________________
Mne_analysis mailing list
Mne_analysis at nmr.mgh.harvard.edu
https://mail.nmr.mgh.harvard.edu/mailman/listinfo/mne_analysis_______________________________________________
Mne_analysis mailing list
Mne_analysis at nmr.mgh.harvard.edu
https://mail.nmr.mgh.harvard.edu/mailman/listinfo/mne_analysisThe information in this e-mail is intended only for the person to whom it
is
addressed. If you believe this e-mail was sent to you in error and the
contains patient information, please contact the Partners Compliance
HelpLine at
http://www.partners.org/complianceline . If the e-mail was sent to you in
error
but does not contain patient information, please contact the sender and
properly
dispose of the e-mail.
-------------- next part --------------
An HTML attachment was scrubbed...
URL: http://mail.nmr.mgh.harvard.edu/pipermail/mne_analysis/attachments/20140324/1a6aacd8/attachment.html
hi Peng,
Thank you Alex for the reply.
1. I suppose you meant freeview is not the optimal tool for visualizing
source space of mne. However, I wish to check the source space by
visualisation before I move on to source reconstructions. I tried
mne_analyze instead of freeview:
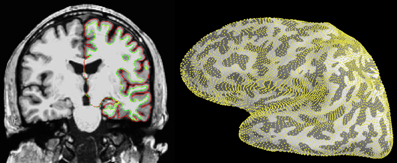
a source space contains the indices of the vertices used in the surface
segmented by freesurfer.
See fig on the right in:

in the recent paper on MNE-Python:
so if you want to check the source space I guess you want to check the
freesurfer segmentation.
1.1 It seemed to detect surfaces generated by freesurfer (such as inflated
etc.) automatically, which I can load by <load surfaces...> in the menu; but
not those surfaces generated by watershed, even after I copy them to the
<surf> subfolder under the subject folder.
watershed is for the BEM not the source space.
you should be able to view them with freeview though.
1.2 I cannot load -src.fif either in mne_analyze. It complains 'Come on, It
is not an MEG/EEG measurement file'...
yes it's not really meant to be visualized. I can send you script used
to make the figure on the frontiers paper if you want.
2. I tried the example in the script you provided (watershed), error
happened while Decimating the dense tessellation..."/Applications/MNE-2.7.4-3378-MacOSX-x86_64/bin/mne_make_scalp_surfaces:
line 125: -nojvm: command not found". I do have <mne_make_scalp_surfaces>
file in the path. (Platform: MacOS10.9.2, macbook pro).
it's because mne_make_scalp_surfaces used matlab for decimation.
there is a python alternative.
Try
mne make_scalp_surfaces
if you have mne python installed correctly.
but it's just for visualization not computation.
3. If my data are MEG instead of EEG, do I still need to use this skin,
skull, brain segmentations from watershed or mne_setup_source_space on brain
surfaces is already sufficient?
you need the inner skull. This command should do the job:
mne_setup_forward_model --homog --surf --ico 4
then mne_do_forward_solution
see :
for a full script.
maybe:
http://martinos.org/mne/dev/command_line_tutorial.html#command-line-tutorial
this can help too although it's not really up to date (but it should work)
Thank you very much and sorry about so many questions...
no pb
Alex
Thanks for the answer, Denis.
Thank you alex.
hi Peng,
> Thank you Alex for the reply.
> 1. I suppose you meant freeview is not the optimal tool for visualizing
> source space of mne. However, I wish to check the source space by
> visualisation before I move on to source reconstructions. I tried
> mne_analyze instead of freeview:a source space contains the indices of the vertices used in the surface
segmented by freesurfer.
I misunderstood it before, I thought it contained were 3-D coordinates.
See fig on the right in:
http://www.frontiersin.org/files/Articles/70133/fnins-07-00267-r2/image_m/fnins-07-00267-g003.jpg
in the recent paper on MNE-Python:
http://journal.frontiersin.org/Journal/10.3389/fnins.2013.00267/full
so if you want to check the source space I guess you want to check the
freesurfer segmentation.I will try.
> 1.1 It seemed to detect surfaces generated by freesurfer (such as
inflated
> etc.) automatically, which I can load by <load surfaces...> in the menu;
but
> not those surfaces generated by watershed, even after I copy them to the
> <surf> subfolder under the subject folder.watershed is for the BEM not the source space.
you should be able to view them with freeview though.> 1.2 I cannot load -src.fif either in mne_analyze. It complains 'Come on,
It
> is not an MEG/EEG measurement file'...yes it's not really meant to be visualized. I can send you script used
to make the figure on the frontiers paper if you want.
Thanks, it will help.
> 2. I tried the example in the script you provided (watershed), error
> happened while Decimating the dense tessellation...
>
> "/Applications/MNE-2.7.4-3378-MacOSX-x86_64/bin/mne_make_scalp_surfaces:
> line 125: -nojvm: command not found". I do have <mne_make_scalp_surfaces>
> file in the path. (Platform: MacOS10.9.2, macbook pro).it's because mne_make_scalp_surfaces used matlab for decimation.
there is a python alternative.
Try
mne make_scalp_surfaces
if you have mne python installed correctly.
but it's just for visualization not computation.
OK, I will double check my Matlab routes and try Python if it still doesn't
work.
> 3. If my data are MEG instead of EEG, do I still need to use this skin,
> skull, brain segmentations from watershed or mne_setup_source_space on
brain
> surfaces is already sufficient?you need the inner skull. This command should do the job:
mne_setup_forward_model --homog --surf --ico 4
Does that mean I will set my source space on inner skull? Would you think
it better on the surface of white matter better or practically no much
difference?
then mne_do_forward_solution
see :
https://github.com/mne-tools/mne-scripts/blob/master/sample-data/run_meg_tutorial.sh
for a full script.
maybe:
http://martinos.org/mne/dev/command_line_tutorial.html#command-line-tutorial
this can help too although it's not really up to date (but it should work)
Great, I will try them then. many thanks again.
> Thank you very much and sorry about so many questions...
no pb
Alex
_______________________________________________
Mne_analysis mailing list
Mne_analysis at nmr.mgh.harvard.edu
Mne_analysis Info PageThe information in this e-mail is intended only for the person to whom it
is
addressed. If you believe this e-mail was sent to you in error and the
contains patient information, please contact the Partners Compliance
HelpLine at
MyComplianceReport.com: Compliance and Ethics Reporting . If the e-mail was sent to you in
error
but does not contain patient information, please contact the sender and
properly
dispose of the e-mail.
-------------- next part --------------
An HTML attachment was scrubbed...
URL: http://mail.nmr.mgh.harvard.edu/pipermail/mne_analysis/attachments/20140325/d3639097/attachment.html
Hi alex,
Some additional questions.
1 After I ran *mne_setup_forward_model, *I have no feedback from the
command line, is it normal?
2 I am using CTF data, thus I converted the .ds folder from CTF to fif
format with *mne_ctf2fiff* command, with option --infoonly. Here I only
want to get the Leadfield thus I suppose data is not necessary. However
when I try *mne_do_forward_solution, *it asked for mri description file,
which I haven't. I have only a series of dicom files or the .mri file
generated by ctf software from them. Can I still move on?
Thanks.
yes it's not really meant to be visualized. I can send you script used
to make the figure on the frontiers paper if you want.Thanks, it will help.
> 2. I tried the example in the script you provided (watershed), error
Trymne make_scalp_surfaces
if you have mne python installed correctly.
but it's just for visualization not computation.
OK, I will double check my Matlab routes and try Python if it still doesn't
work.> 3. If my data are MEG instead of EEG, do I still need to use this skin,
> skull, brain segmentations from watershed or mne_setup_source_space on
> brain
> surfaces is already sufficient?you need the inner skull. This command should do the job:
mne_setup_forward_model --homog --surf --ico 4
Does that mean I will set my source space on inner skull? Would you think it
better on the surface of white matter better or practically no much
difference?
there is 2 different and independent things here:
- source space
- surfaces for forward model
skull is for the forward model and has nothing to do with the source space.
HTH
A
hi,
Some additional questions.
1 After I ran mne_setup_forward_model, I have no feedback from the command
line, is it normal?
it should print that it saved a file to disk
2 I am using CTF data, thus I converted the .ds folder from CTF to fif
format with mne_ctf2fiff command, with option --infoonly. Here I only want
to get the Leadfield thus I suppose data is not necessary. However when I
try mne_do_forward_solution, it asked for mri description file, which I
haven't. I have only a series of dicom files or the .mri file generated by
ctf software from them. Can I still move on?
you need to do the coregistration and get a -trans.fif file.
A
Thank you for the answers.
1. Surprisingly I did not find any files were generated/saved after running
mne_setup_forward_model.
2. I don't have a -trans.fif file. I tried to do it in GUI of MNE_analyze,
it was quite complicated. If I have a co-registered mri file (generated by
CTF software with .mri extension), can it be converted to fif format? If
not, I can read the 4x4 head2mri matrix via matlab, can this information be
written to fif format with certain tools in MNE?
Thanks again!
Thank you for the answers.
1. Surprisingly I did not find any files were generated/saved after
running mne_setup_forward_model.
I assume there was an error, can you post the program output?
2. I don't have a -trans.fif file. I tried to do it in GUI of
MNE_analyze, it was quite complicated. If I have a co-registered mri
file (generated by CTF software with .mri extension), can it be
converted to fif format? If not, I can read the 4x4 head2mri matrix via
matlab, can this information be written to fif format with certain tools
in MNE?
There is also a coreg GUI in MNE-Python, have a look here
You could write a .fif file with the transform in Matlab, but it seems
to me that this wouldn't be easier than using the coregistration tools
in mne_analyze or MNE-Python.
HTH,
Martin
Thank you Martin for the help.
1. I re-run the command "mne_setup_forward_model" and it seemed working
this time (maybe I did not set the environment correctly). Sorry for the
confusion.
2. I tried with MNE_analyze GUI and it was not easy for me. But I saved
results from one subject successfully and it could be load by MNE_analyze.
However I wanted to read it in matlab to check the real contents (which I
assume is a structure with the transformation matrix). I failed with "x =
fiff_read_mri(fname, 0). It complained "Could not find MRI data"... Would
please let me know the function name to read and write *-src.fif file in
matlab?
Dear Alex, Martin and other MNE users,
I have my MEG raw data in CTF .ds format and raw MRI image with dicom
format. I wish to use MNE to compute the leadfield (source space based on
surface).
With your help, I wish to summarise a possible pipeline as a following:
1. recon-all --subject test1 #use in freesurfer to generate the brain
surfaces and other needed files.
2. mne_watershed_bem --subject test1 --overwrite --atlas #generate surfaces
for bem model
3. mne_ctf2fiff --ds test1.ds --fif test1.fif --infoonly #generate MEG
information (sensor locations etc.)
4. #use MNE_analyze with test1.fif and inflated.surf to generate
test1-trans.fif which contains the transformation matrix;
#or use a previous calculated transformation matrix and convert it to
fif format
5. mne_setup_source_space --subject tmp --ico 5 --morph test1 #tmp is name
of a template, could be fsaverage or icbm152...
#This is to use locations on the surface of the template cortex (white
matter?) as source space.
#These locations was morphed to subject <test1> to generate leadfield for
this subject later
6. mne_do_forward_solution --subject test1 \
--src test1-fsaverage-ico-5-src.fif \
--meas test1.fif \
--trans test1-trans.fif \
--megonly --overwrite \
--fwd test1-oct-5-fwd.fif
# I can then find my leadfield (MxNx3 matrix, M=number of locations, N =
number of sensors) by importing test1-oct-5-fwd.fif into matlab.
Did I miss something?
Thanks a lot for the help.
Sorry, the last line should be read and write <*-*trans*.fif> file in
matlab?
That certainly is the basic workflow in terms of commands, but I don't
think you want to use the "tmp" subject. You also don't morph the data
until later in the process.
Can I recommend that you start off by doing the example in the
"Cookbook" chapter? It does not take very long, and it will help you
be more familiar with how the process works in one individual. Then
you can try to take what you learn from that and apply it directly to
your data.
HTH,
D
.
Hi Peng,
The matlab toolbox chapter in the manual specifies how you read each
file type. The function to read a "-src.fif" file is
mne_read_source_spaces.
D
Hi Peng,
Please read carefully chapter 12 of the manual, and you will master how to
process MEG data. After you know it, you can try to process data following your
imagination. Of course, each time you encounter problems, please read the
associated chapters of the manual.
Best wishes,
Junpeng
2014-03-28
junpeng.zhang
???dgw <dgwakeman at gmail.com>
???2014-03-28 22:08
???Re: [Mne_analysis] a junior question on mne_setup_source_space
???"Discussion and support forum for the users of MNE Software"<mne_analysis at nmr.mgh.harvard.edu>
???
That certainly is the basic workflow in terms of commands, but I don't
think you want to use the "tmp" subject. You also don't morph the data
until later in the process.
Can I recommend that you start off by doing the example in the
"Cookbook" chapter? It does not take very long, and it will help you
be more familiar with how the process works in one individual. Then
you can try to take what you learn from that and apply it directly to
your data.
HTH,
D
.
Dear Peng,
you have here full scripts to analyze CTF data:
for the anatomy pipeline:
https://github.com/mne-tools/mne-scripts/tree/master/spm_face
for MEG data analysis:
http://martinos.org/mne/dev/auto_examples/datasets/plot_spm_faces_dataset.html
HTH
A
Thanks, dgw and junpeng for your feedback. I did read the manual before
asking:)
I am sorry '-src.fif' was a typo, I wanted to read -trans.fif. I did not
find a proper function in the manual.
To write it, <fiff_write_coord_trans> seem to be the correct function.
According to the manual, I set
{kind=link}